Overview
You will now want to set some optional tuning parameters in the XML file that is directly relevant to the desired analysis. Valid analysis strings are "tumor_cnv" and "seq_cnv". The latter analysis is currently under development.
The XML file must be named
Tumor CNV configuration
tumor_cnv.xml expects in one of its field a filename that contains a list of signal intensity input files. This manifest file has a header describing the following columns: the path of the input file, a friendly name that will be pre-pended to the results to identify the dataset, and the stromal contamination level of the sample.
Each input file has a header describing the following columns: the SNP identifier, the chromosome (all records must contain the same chromosome), the base pair position (this must be sorted in increasing numerical order), the B allele frequency, and the log2 R ratio.
Stromal contamination prediction
tumor_purity.pl is a Perl script that can be used to predict the fraction of tumor cells within the sample.
The required input includes a signal intensity file (text file with SNP, Chr, Position, Log R Ratio and B Allele Freq) and a HMM file:
Usage:
tumor_purity.pl [arguments] <input-signal-file> <PennCNV-HMM-file>
Optional arguments:
-v, --verbose use verbose output
-h, --help print help message
-m, --man print complete documentation
--snpposfile <file> a file with chr/position information for markers
--bin <int> the BIN for grouping SNPs together (default: 100)
--grid <int> the GRID for precision of estimate (default: 50)
--portion <float> portion of LRR windows for estimation (default: 0.5)
Function: calculate tumor purity (1-stromal contamination) levels from signal intensity file with LRR/BAF values
Example: tumor_purity.pl signal.txt hhall.hmm
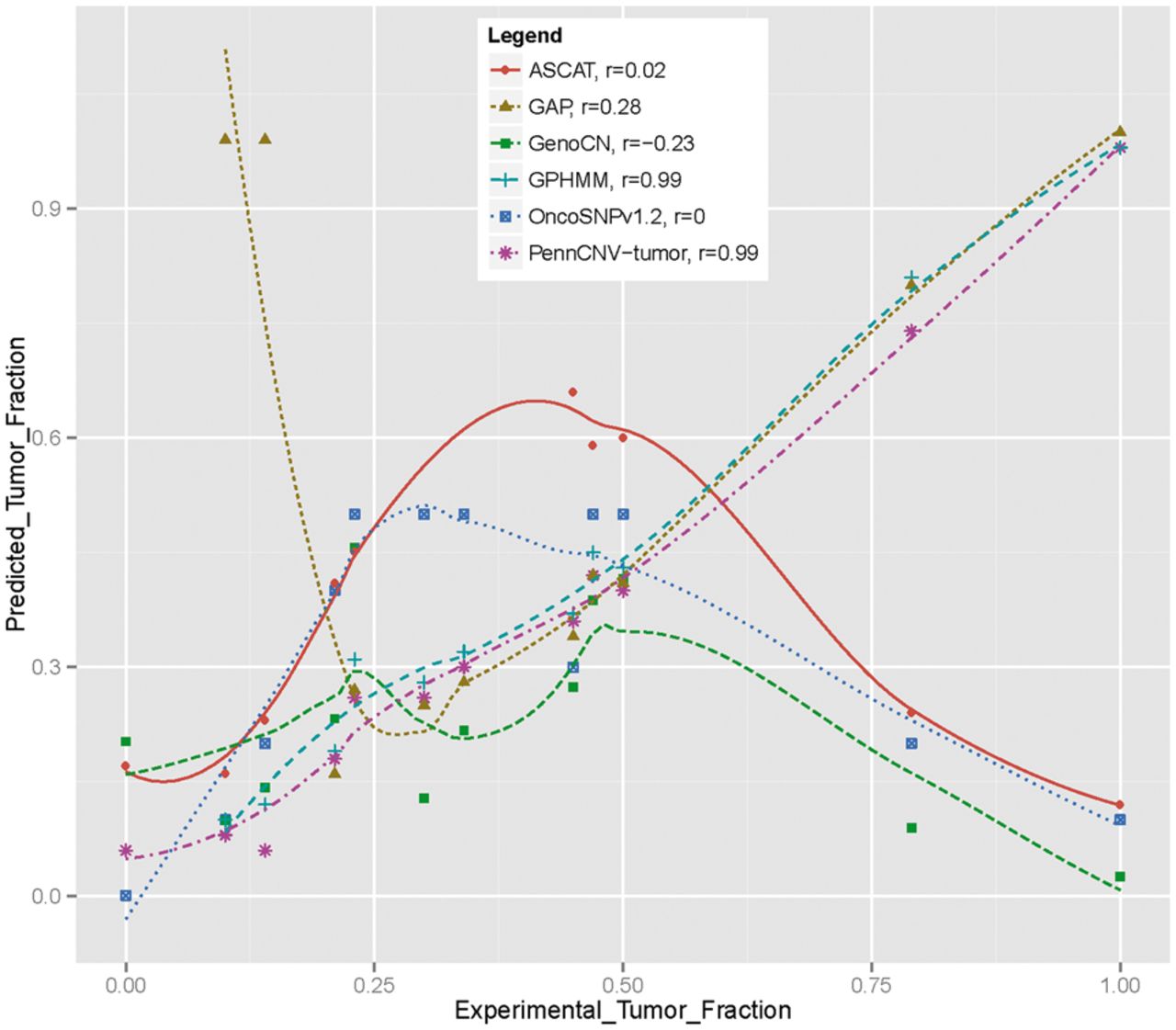
An example is shown below that illustrate the strong correlation between predicted stromal contamination and experimental values.
CNA detection
You can now execute the program by running
./analyzer <analysis string>
An example is shown in examples directory.